![]()
Relative Energies Automated Calculation inTerface
REACT - making computational chemistry easy
REACT is developed by PhD Geir Villy Isaksen and MSc Bente Sirin Barge from UiT The Arctic University of Norway. The main vision behind is to deliver a compact, intuitive & efficient software for energy calculations taylor made for enzyme catalysed reactions.
What it does
- Automatic calculation of relative energies and easy inclusion of typical correctional terms:
- Solvation
- Big basis
- Frequencies / thermal corrections
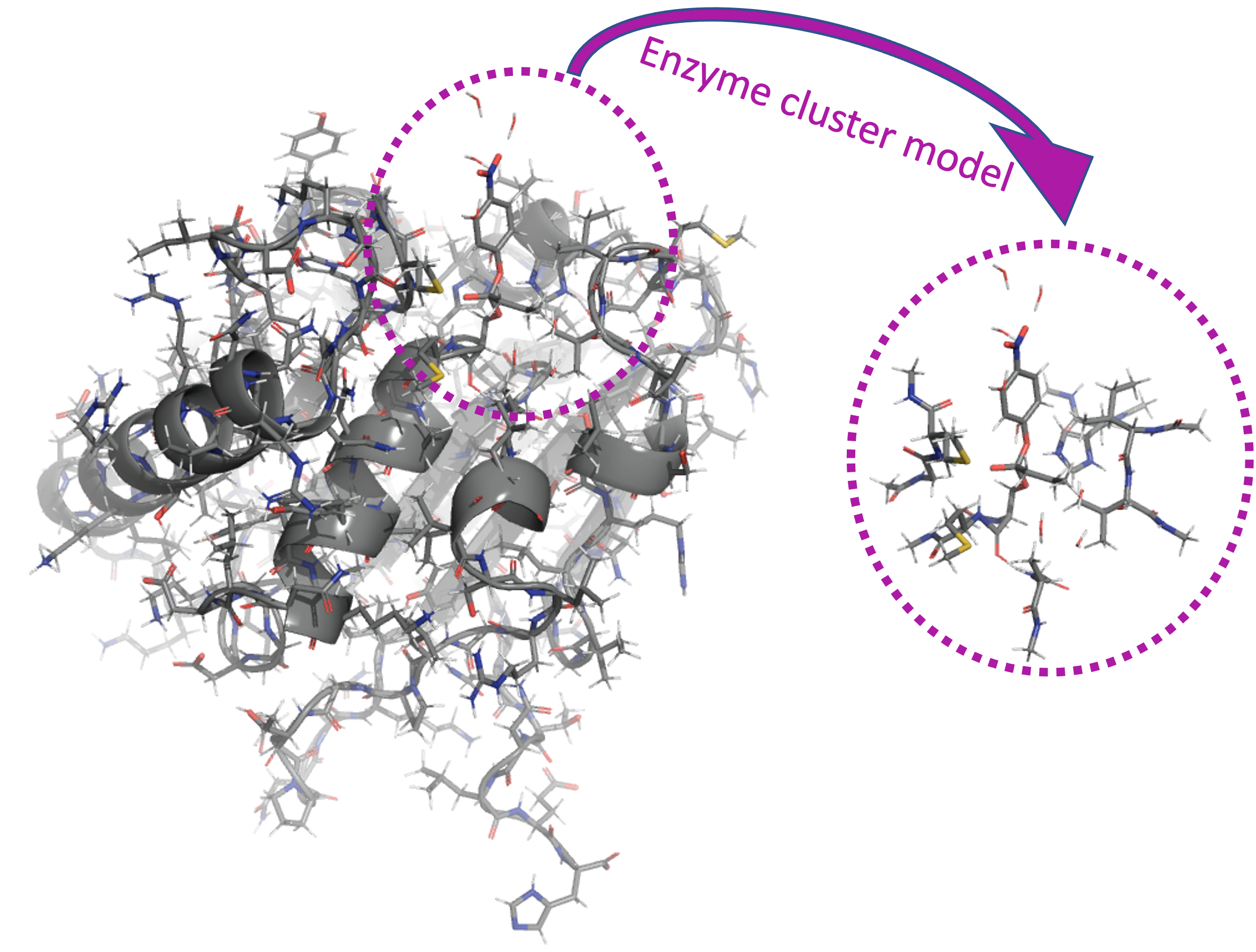
- Generation of enzyme cluster models
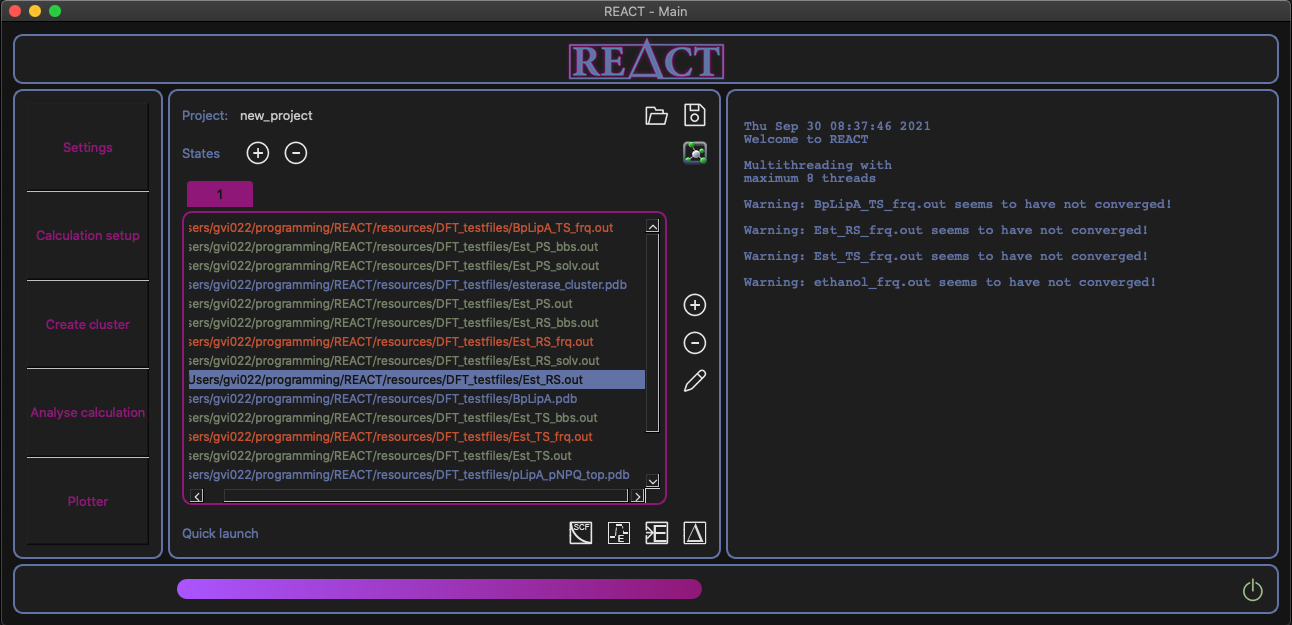
- Store all related project files with respect to where they belong in a reaction pathway (states)
- Handling of several filetypes (pdb, xyz, com, out, inp) in a filetype-independent way
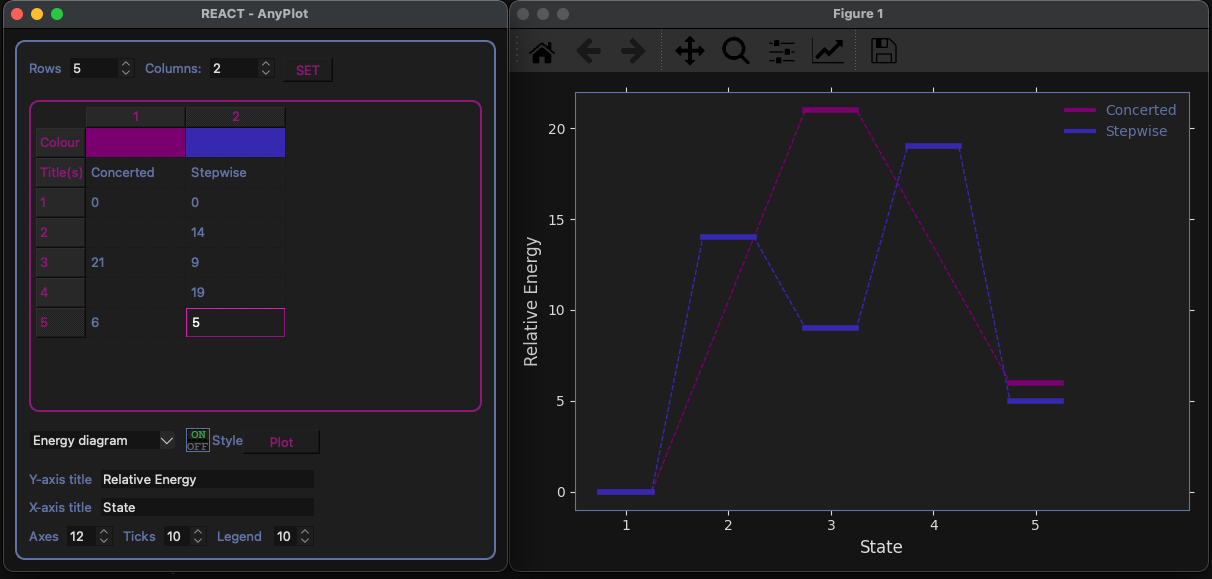
- Plotting functionalities to visualize:
- Energy diagrams
- SCF convergence
- Frequencies / IR spectrum
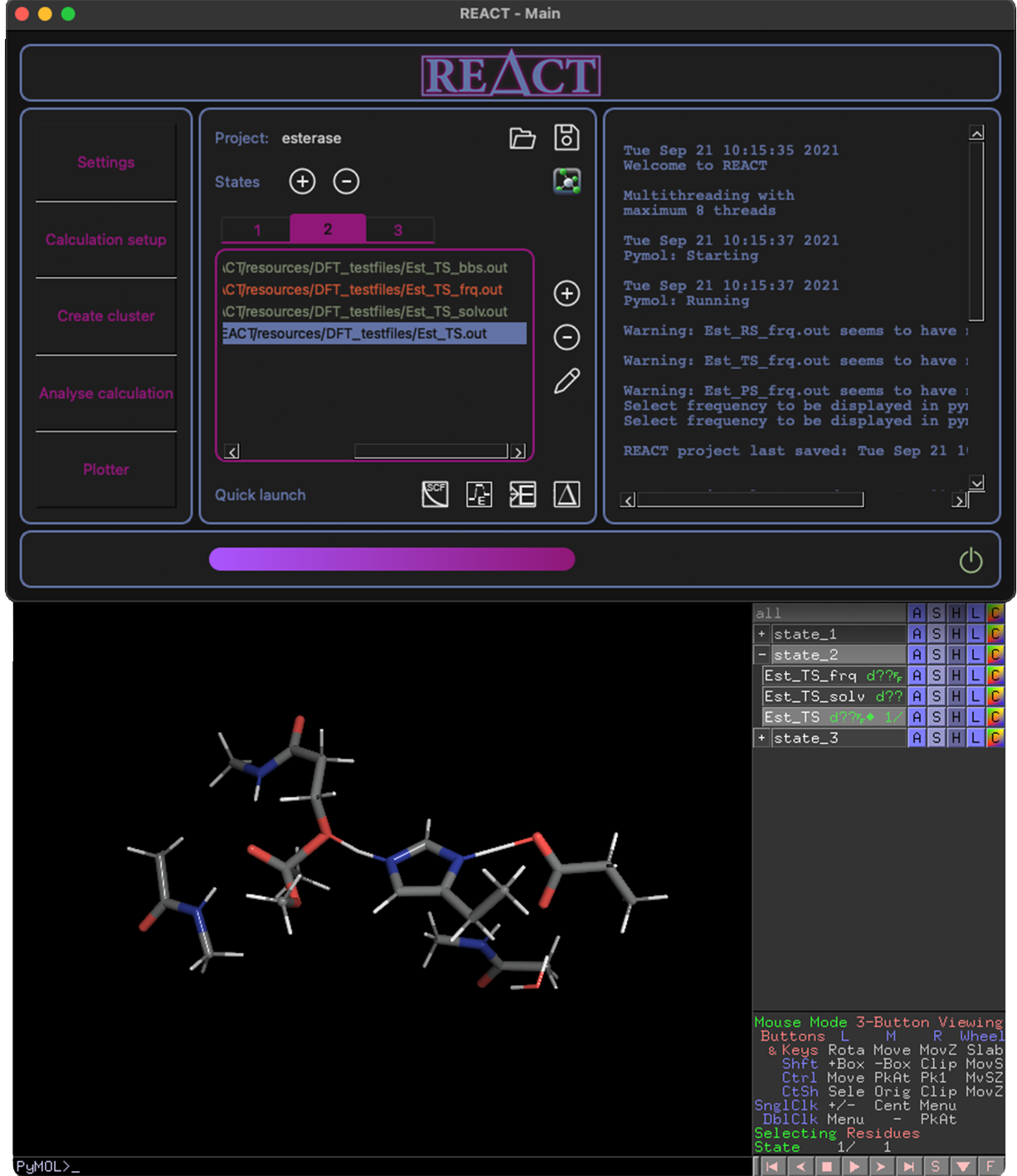
- Pymol connectivity for molecular visualization of all project files
- Frequency animation
- SCF geometry optimization visualisation
- Enzyme cluster model generation
- Easy setup of new input files for DFT/QM calculations
What it does not
- The actual QM/DFT calculations.
There are several excellent software packages on the market for doing hard-core quantum mechanics (QM) and density functional theory (DFT) calculations. Our goal has been to design an efficient project tool that automates many of the tedious tasks required to both set up and post-analyse QM/DFT calculations.
Supported DFT / QM software packages
Currently, we have only implemented input/output support for Gaussian. REACT is however designed with the purpose of easy support implementation for other QM/DFT engines.
Screenshots
REACT main window
All project files and states synced to Pymol
Creating run-ready cluster models
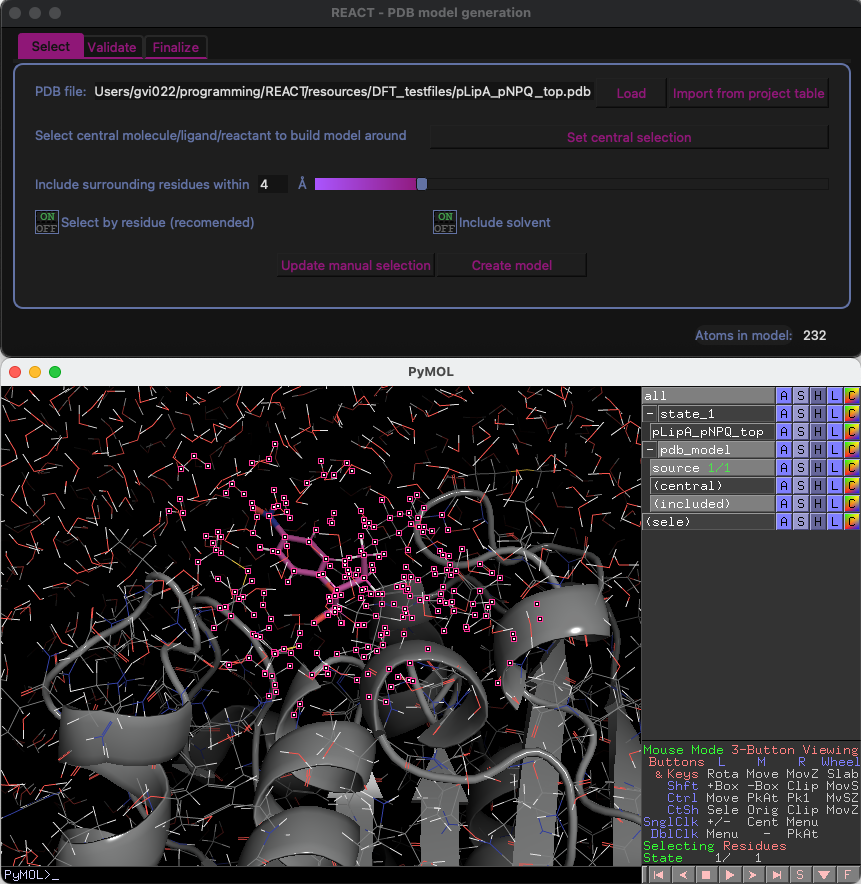
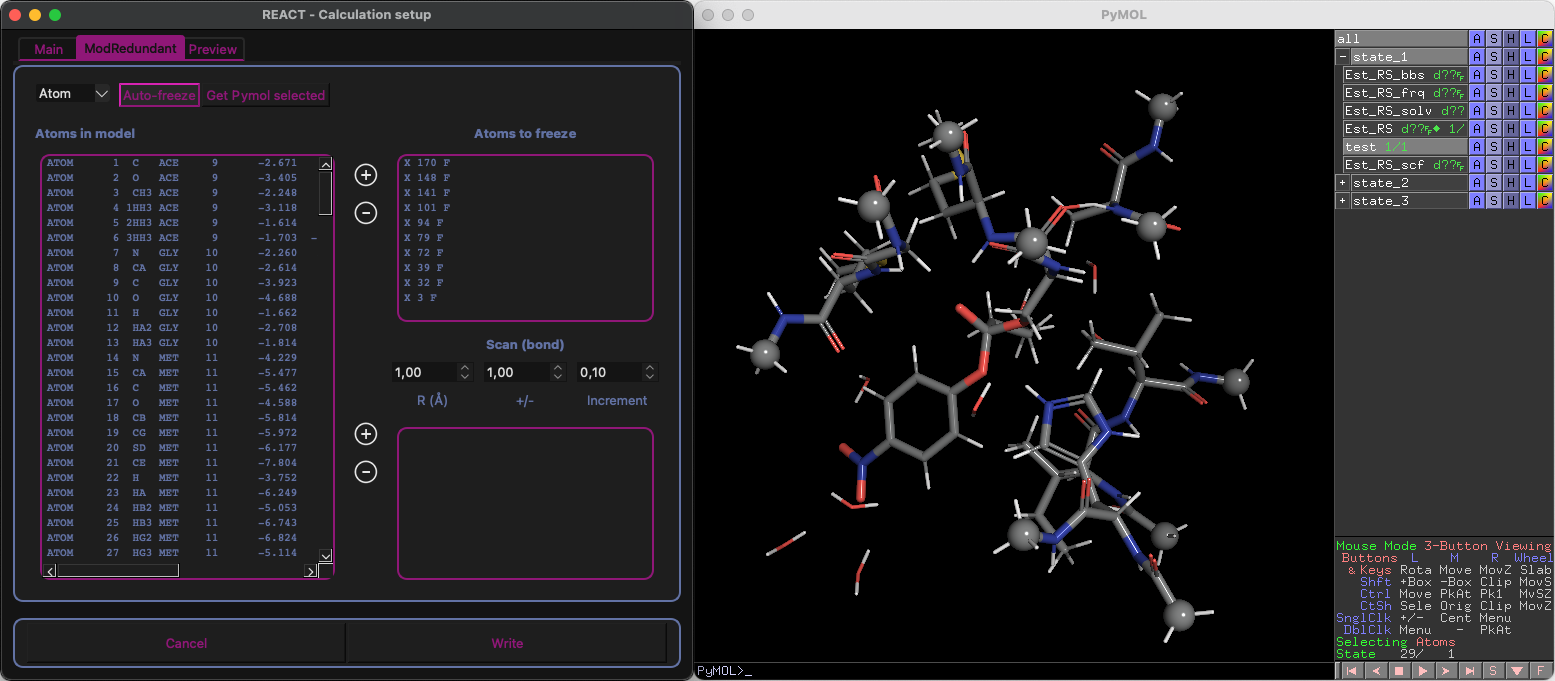
Automatic detection atoms to freeze (cluster)
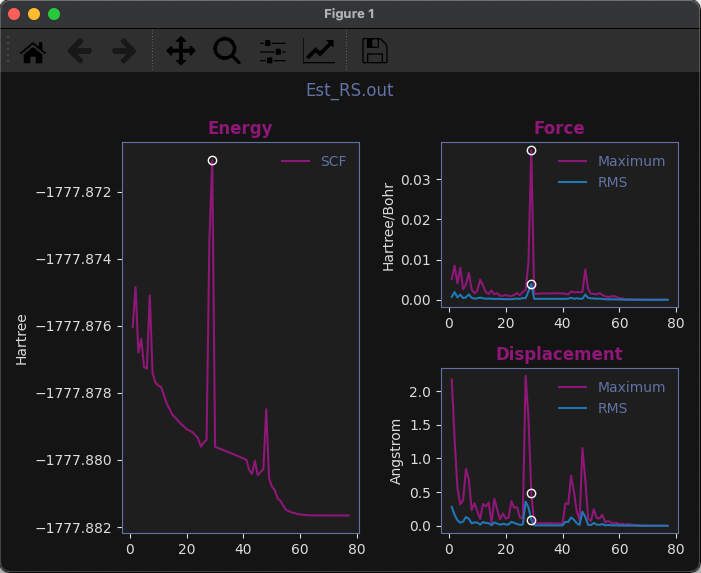
Familiar convergence plots
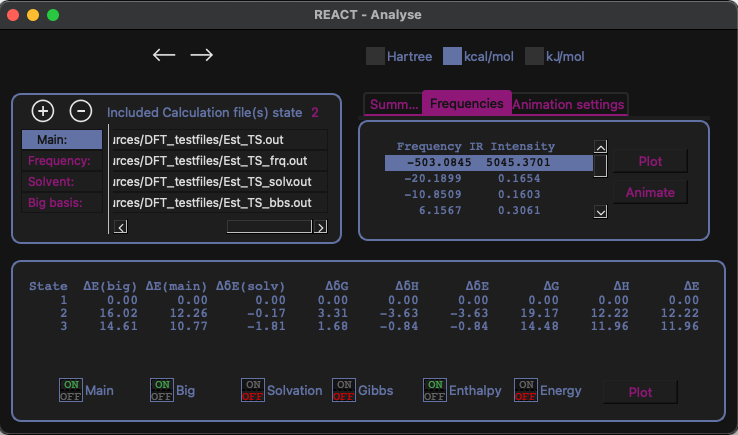
Efficient analysis tool
Energy diagram custom plotting tool